Standard structure and admixture-analyses miss information of populational history by amplifying most common allele linkages
between individuals and by distorting gene flows and flow directions. I have built a new testing installation figuring allele mismatch between test groups. In this test a small mismatch doesn't tell straightly difference or similarity between individuals. Instead of small differences, which actually usually can't be identified by origin and long history, the amount of shared alleles entirely in object population tells the story.
So the result indicates what is entirely common between proposed admixture groups and tested individuals and the test dismisses distinct admixtures. Admixture and Structure, as well as PCA, do the trick conversely, creating a wow-effect if there is minor admixtures or recent genetic drift.
But to avoid the impact of genetic drift the comparison is in my test done using
non-related third reference populations as middlemen. As far as I can tell in case of Finns the calculated allele mismatch matches well with the known history. In other words, distinct admixtures and recent genetic drift are disregarded and the results show a common root of object populations and tested individual.
I have now tested people belonging to my project, excluding Scandinavians. For Scandinavians I'll do another test case, because they need very different admixtures than Finns.
Average allele mismatch figures for Southwest and East Finns:
East Finnish / Sweden 87
East Finnish / West Russia 54
East Finnish / Estonia 29
East Finnish / Karelia 8
East Finnish / Veps 11
East Finnish / Poland 84
Southwest Finnish / Sweden 37
Southwest Finnish / West Russia 23
Southwest Finnish / Estonia 7
Southwest Finnish / Karelia 29
Southwest Finnish / Veps 36
Southwest Finnish / Poland 39
Project members' results:
Equivalences for old project member identity codes
HM0001 = FI1
LS0001 = FI2
LS0002 = FI3
LS0003 = FI4
KA0001 = FI5
LS0004 = FI6
announcement sent = FI7
announcement sent = FI8
announcement sent = FI9
SK0001 = FI11
me = FI12
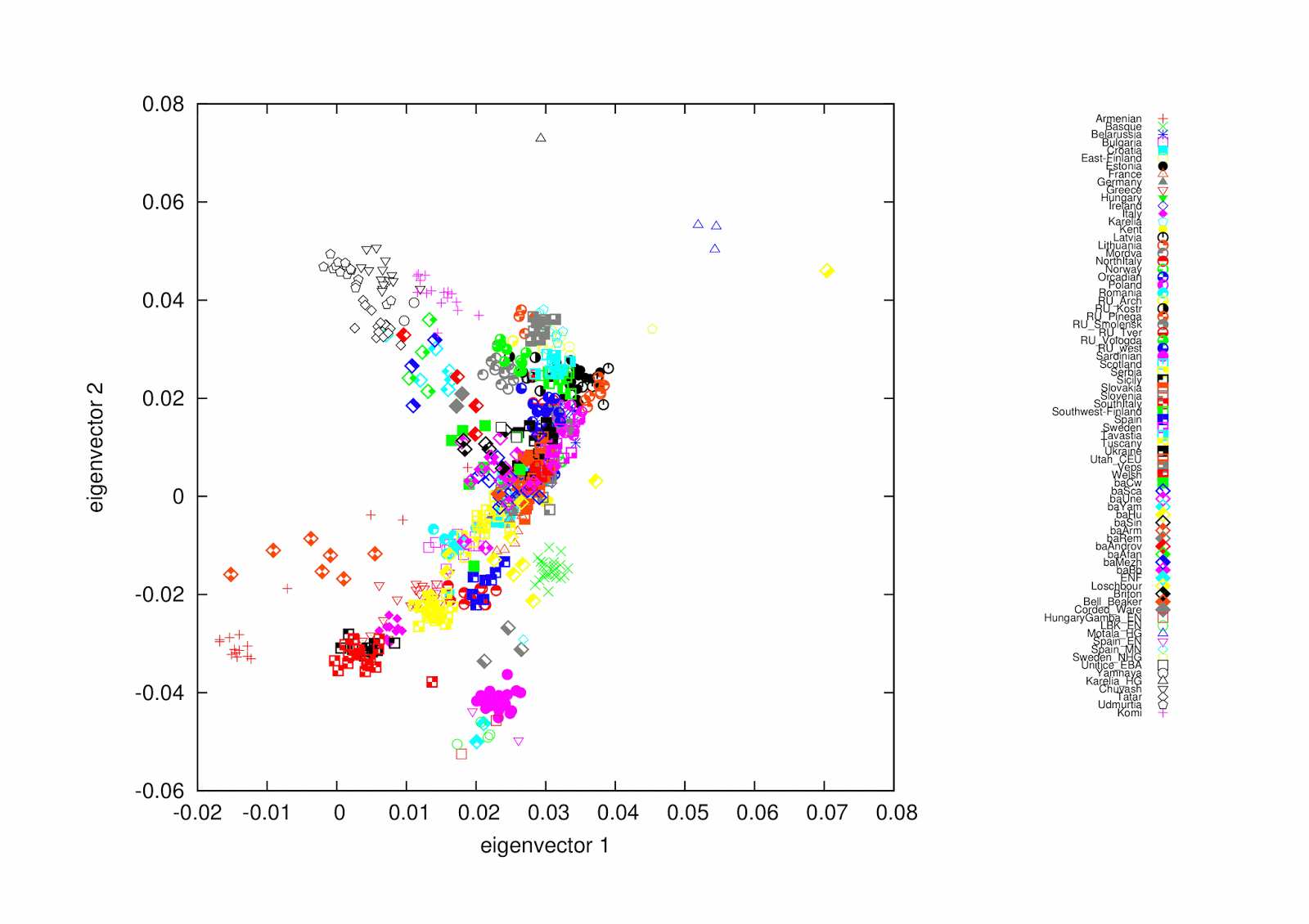
For data checking here is a PCA (look, it smiles):
edit 6.11.2015
Here are results for Swedish project members. Some notes
- SC2 has probably Baltic or Slavic admixture
- SC3 has West Russian and possibly Southwest Finnish admixture
- SC6 has more Finnish admixture
I understand that readers can question those non-Swedish admixtures and I appreciate if SC2, SC3 and SC6 could leave their truthful comments anonymously here.
Something to notice that none of those admixtures can be seen on PCA.