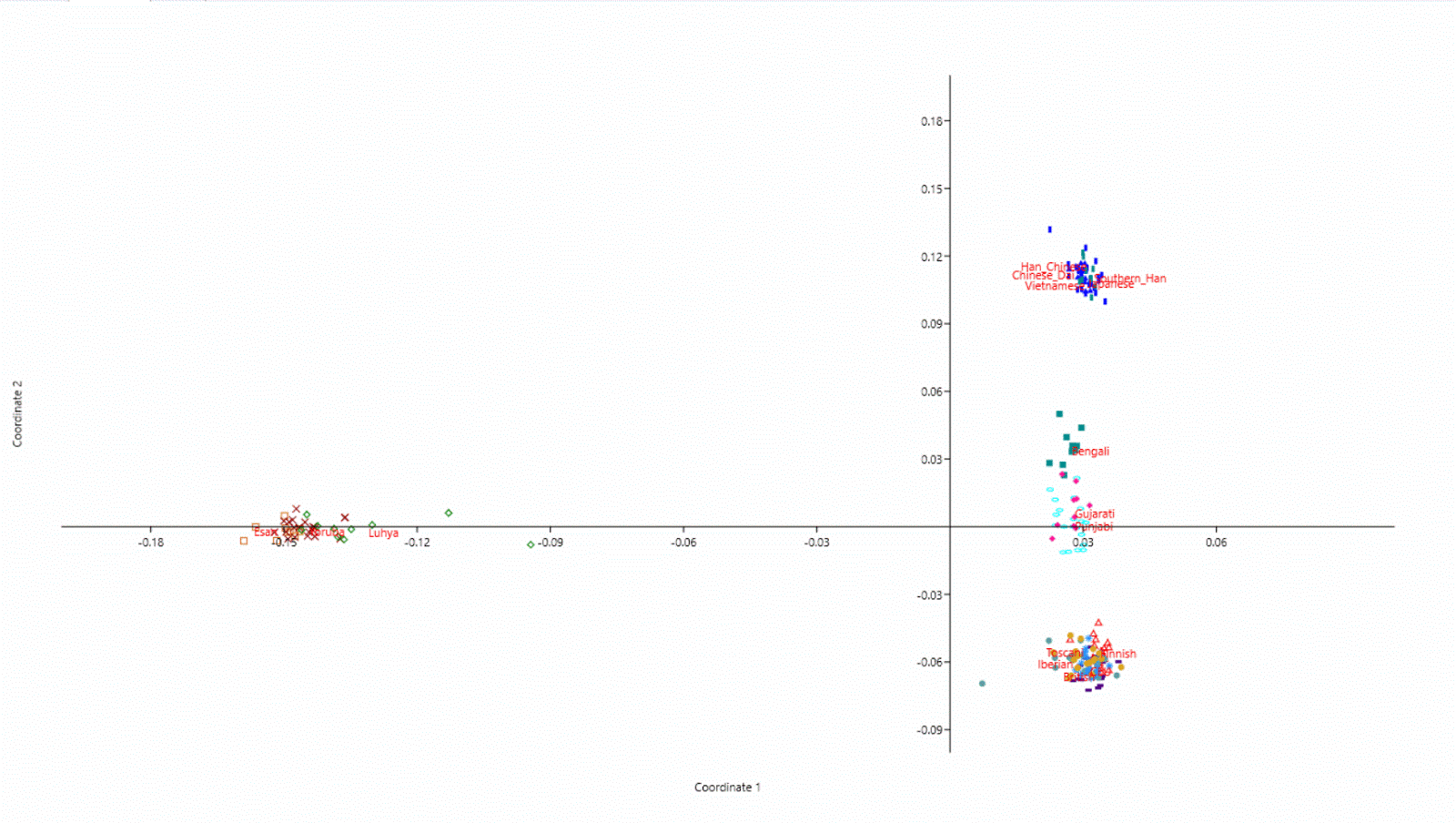
The EGDP data, available from Estonian Biocenter, made it possible to reach 15-30 times more genome density than earlier available data made possible. The new data lacks of West European samples, but it was not a big problem due to the publicly available western data from the 1000-genomes project. So I merged these two data sets. For the quality check I ran heterozygosity rates for all European samples in both data sets and found both sets being considerably close each other, although the read depth of the 1000-genome data is smaller. Actually Finnish samples in both sets showed exactly same level of heterozygosity.
After the succesful merge I had 14.4 million SNPs over all 22 chromosomes, which was far too much to process in few days on my desktop (i7, 3.5Ghz, 32 GB memory). Instead of thinning the whole data set to 1-2 millions SNPs I decided to use chromosomes 1 and 6 and leave the genome density untouched. So I had two chromosomes, a bit over 2 million SNPs showing still 15-30 times more genotype information per chromosome than other available genotype sets. Considering thinning over all chromosomes to get the dataset handy enough to be processed with my computer would likely have induced more algorithm dependent bias, which I wanted to avoid.
The process
1 merging EGDP and 1000g data sets
2 quaility checks, including homozygosity/heterozygosity ratios per populations
3 extracting chromosomes 1 and 6
4 thinning data by Plink: plink --file data --indep 50 5 2, resulting 1.1 million SNPs
5 running admixture analyses with k values from 3 to 13 in
unsupervised mode and without reference populatons (=projection).
Each k-value was run in unsupervised mode without reference data, because
projection reference data is not available for this SNP set. You can see analyses using
projection reference for example in works analysing ancient and moderm genomes together. Analyses made on any kind of projection are cool, because we have no other way to designate proportion of ancient samples to modern ones. I am not saying that
unsupervised analysis without references would be error-free, but that errors are systemic and not user dependant.
All analyses (k-values from 3 to 13) done here are run as individual runs without user supervision and for that reason colors on charts are not consistent (at least it sounded like a painful work the get colors consistent). Each analysis is optimized separately by the Admixture algorithm. All this makes it more difficult to perceive differences between different K values, but as soon as you get the idea I am sure you also can see the big picture and understand details.
Hopefully this test is helpful for you. In my opinion, it gives interesteing hints about Finnish relations with other populations, but the analysis itself is wordwide.
- Mordvins seem to differ from other Volga-Finnic populations and belong to Balto-Slavic ancestry and they probably are language shifters from a Baltic to a Volga-Finnic language.
- Estonians are just what can be expected, some Estonians have Baltic ancestry, some others Baltic-Finnic ancestry. We should, however, be cautious of in using linguistic terms when we speak about ancestry.
- North Russian Finno-Ugric populations seem to be Baltic-Finnic people with Siperian admixture. The Siberian admixture is present in a lesser amount among Finns and Estonians (note that the amount of minor admixtures depends on the used data/populations and Admixture is based on a selective method processing admixture proportions relatively).
- in some extent also Swedes show Baltic-Finnic ancestry, but the Swedish sample size is rather small to make a sure conclusion. However,
if this is true, we can assume the present-day Baltic-Finnic people having largely Fennoscandinavian ancestry.
- Ingrian samples show up like pure unadmixed Baltic-Finnic people, which surprises me because of their long lasting minority status in Russia. Sample collectors have done good work. Those samples are valuable indeed.
- thinking all this and trying to rebuild the the history of Baltic-Finnic people it looks like they lived to the north from the axis Latvia-Moscow (Balts living to the south before the East-Slavic expansion). Mixing between Baltic and Finnic people happened and people also shifted language.
- open questions are how strong the Baltic-Finnic influence is/was in Scandinavia and conversely how strong the Germanic influence is/was in Finland and Estonia. For certain political reasons it is a difficult approach today.
CV errors, indicating quality in general, the lower the value is the better the quality, but absolute values depend on the used data and can't be compared to other Admixture tests.
K3: 0.19708
K4: 0.19503
K5: 0.19480
K6: 0.19451
K7: 0.19432
K8: 0.19503
K9: 0.19508
K10: 0.19576
K11: 0.19708
K12: 0.19797
K13: 0.20221
Population abbreviations, download
here
Analysis, download
here.
You definitely need a suitable picture viewer being able to handle big GIF-files.






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}