I made this test using Chromopainter and Finestructure. Unfortunately Chromopainter is a rather ineffective tool and incapable to use available computing resources (threads, memory). Without this drawback I would have made this using 25 millions markers instead of only 3.2 millions.
The process:
1 Vcftools, parameters -remove indels -chr 23
2 Haplytyping using HAPI-UR and all samples, run three times and driven in consensus
3 Made a manual selection for random samples, 10-20 of each population
4 Chromopainter, without specifying donor haplotypes
5 Finestructure with run parameters 30000/300000
6 MDS using Past.
Additionally I ran Vcftools using parameters -keep-only-indels and -chr 23. The result was filtered and biallelic deletions (CN=0) were counted. Male results were treated biallelic, so CN=0 should give us the number of effectine deletions in both cases, for females and males.
Finestructure





MDS done by Past:
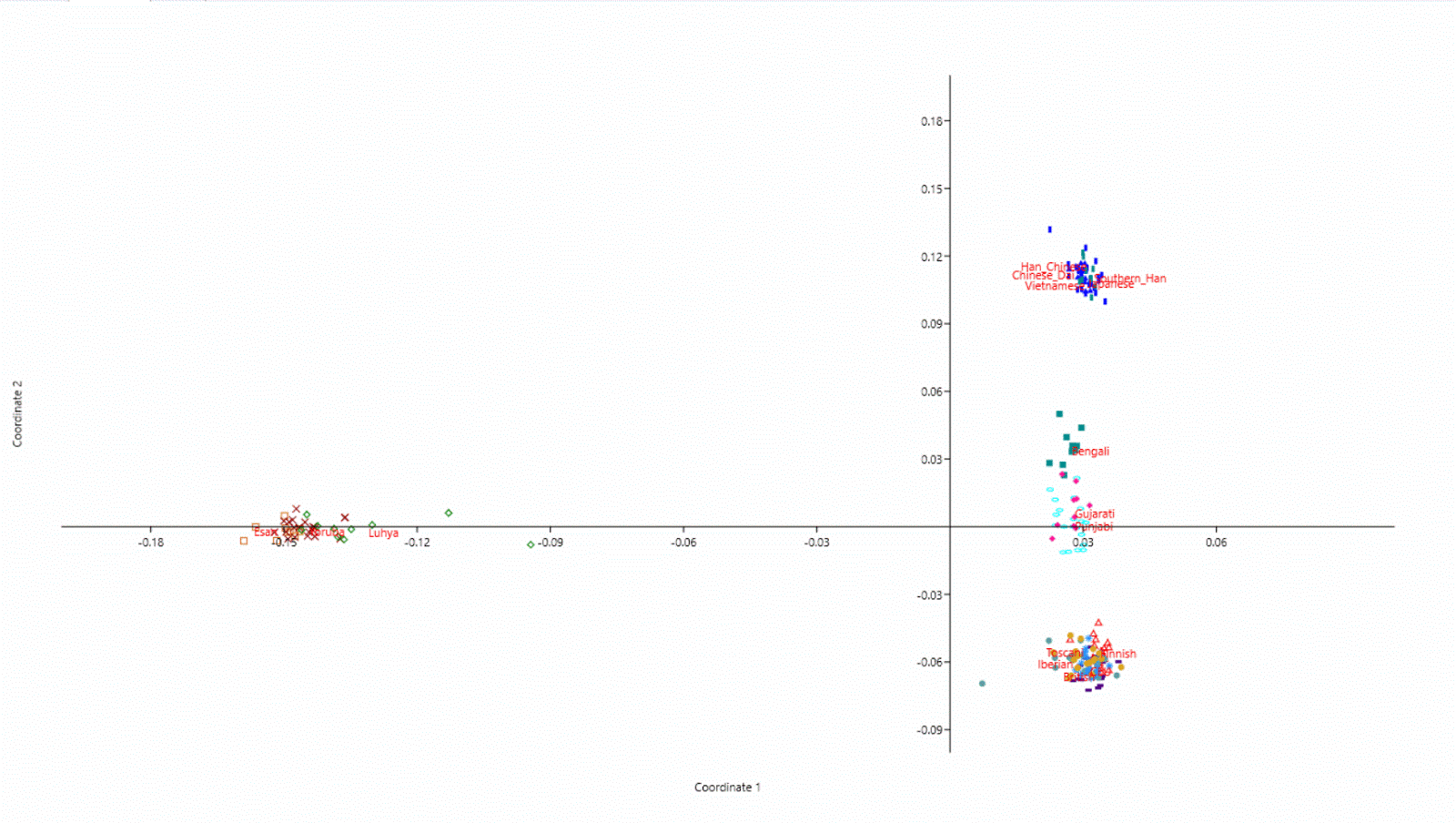
All previous pictures are downloadable with better resolution, here.
Deletions per 3.2 million markers (averages per sample):



The British subgrouping is gathered from internet and can be unreliable. The Finnish one represents those with highest Siberian admixture, the group being "most Finnish" / local, those closest ancient Corded Ware samples and the rest of all 99 samples. The last Finnish group includes all outliers.